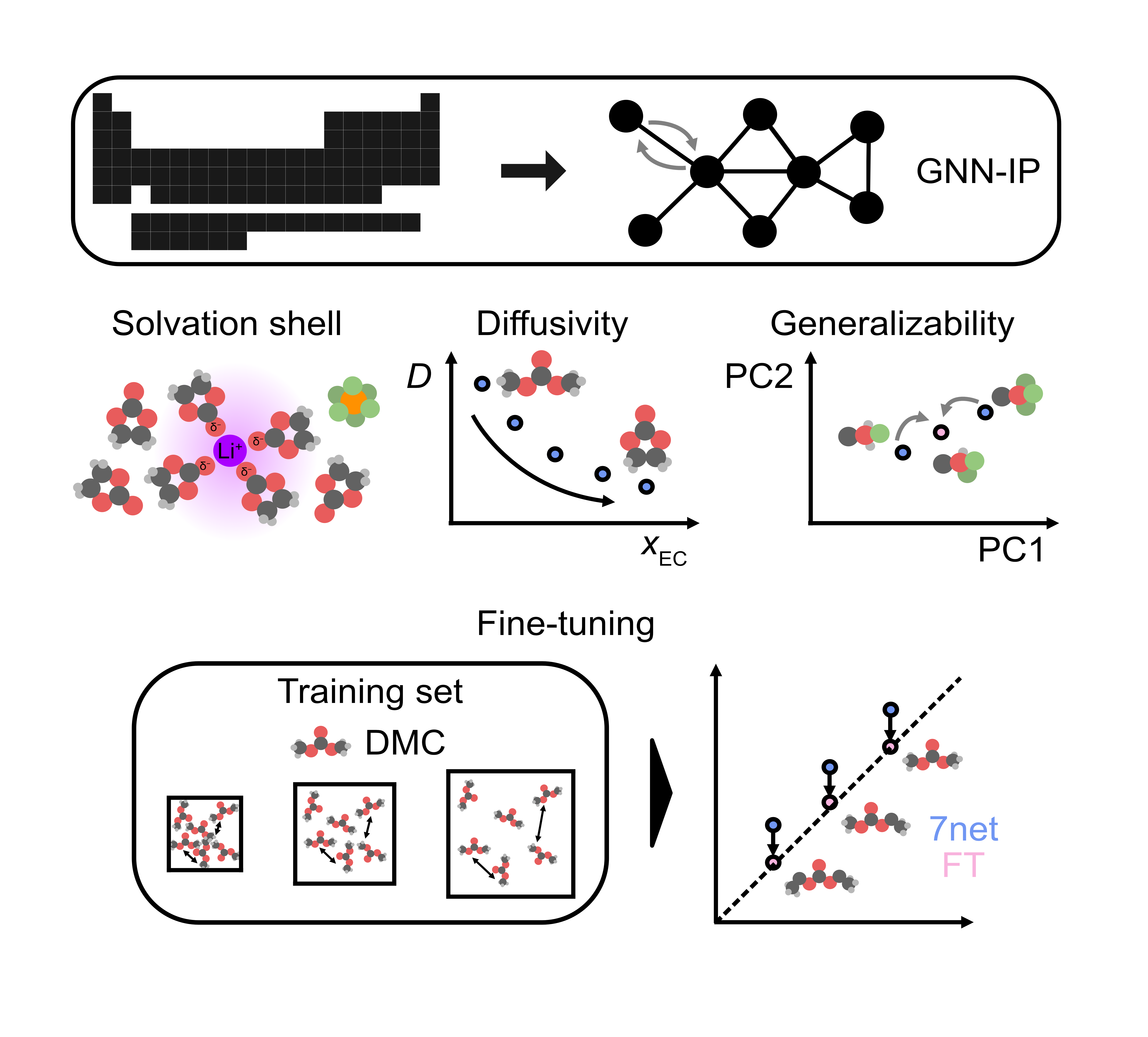
Improving Li-ion batteries to support higher voltages, faster charging, and wider temperature ranges requires better electrolyte materials, but exploring the vast combinations of solvents, salts, and additives is challenging using traditional experimental and computational methods. Here, we evaluated a state-of-the-art pretrained universal machine-learning interatomic potential (MLIP), SevenNet-0, for its ability to predict key properties of liquid electrolytes, including solvation structure, density, and ion transport. Although primarily trained on inorganic systems, SevenNet-0 performed well on organic electrolytes, showing good agreement with experimental and ab initio data, though we identified systematic errors, particularly in density predictions, and addressed them by fine-tuning the model. This approach improved accuracy significantly while keeping computational costs low, and analysis of the training set indicated that the model achieved its performance by generalizing across chemical space rather than memorizing specific configurations. This study highlights the potential of MLIPs like SevenNet-0 to accelerate the discovery and design of advanced electrolyte materials, and this work was published in Digital Discovery 2025, 4, 1544.