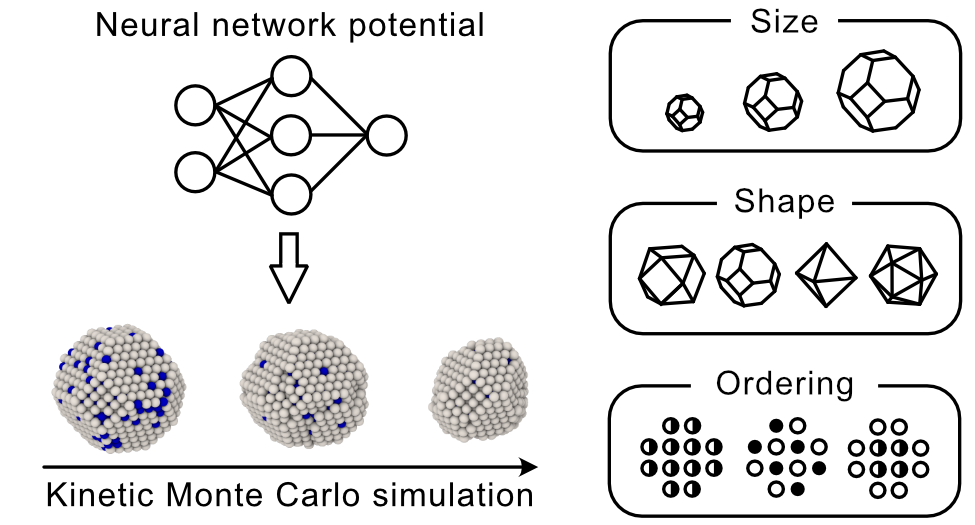
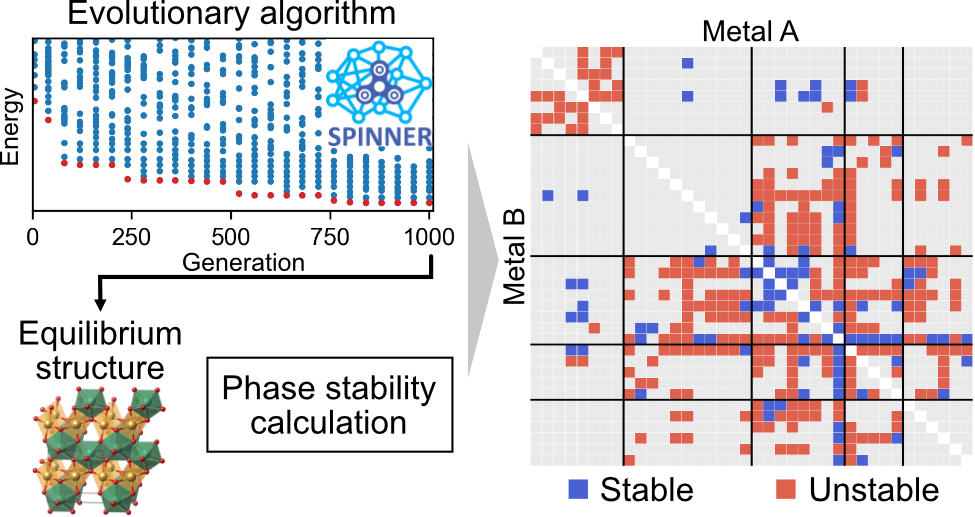
Ternary metal oxides have played a crucial role in advancing technologies, but the low throughput of experimental synthesis hinders the discovery of new oxides. In this study, we utilize SPINNER, which combines neural network potential and evolutionary algorithm, to scan 181 ternary metal oxide systems that have not yet been reported and discover 45 systems containing novel oxides that are more stable than related binary compounds or elemental phases. By directly searching up to 60,000 structures, SPINNER overcomes the limitations of material discovery with data mining methods that only search for prototype structures. This work paves the way for more accurate discovery of new multielement compounds that can be synthesized prior to experimentation at modest computational cost. This study was published in Journal of the American Chemical Society, 2023, 145, 35, 19378–19386