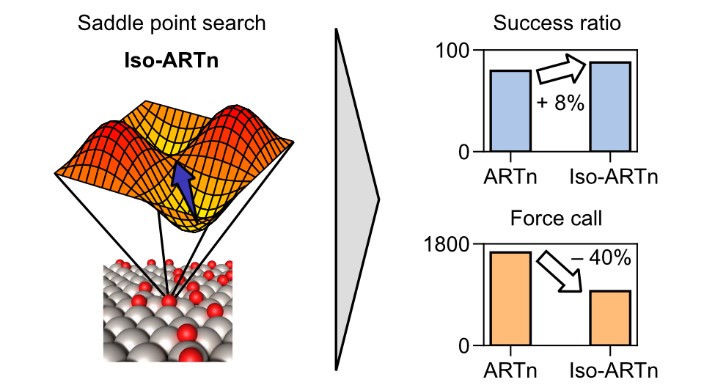
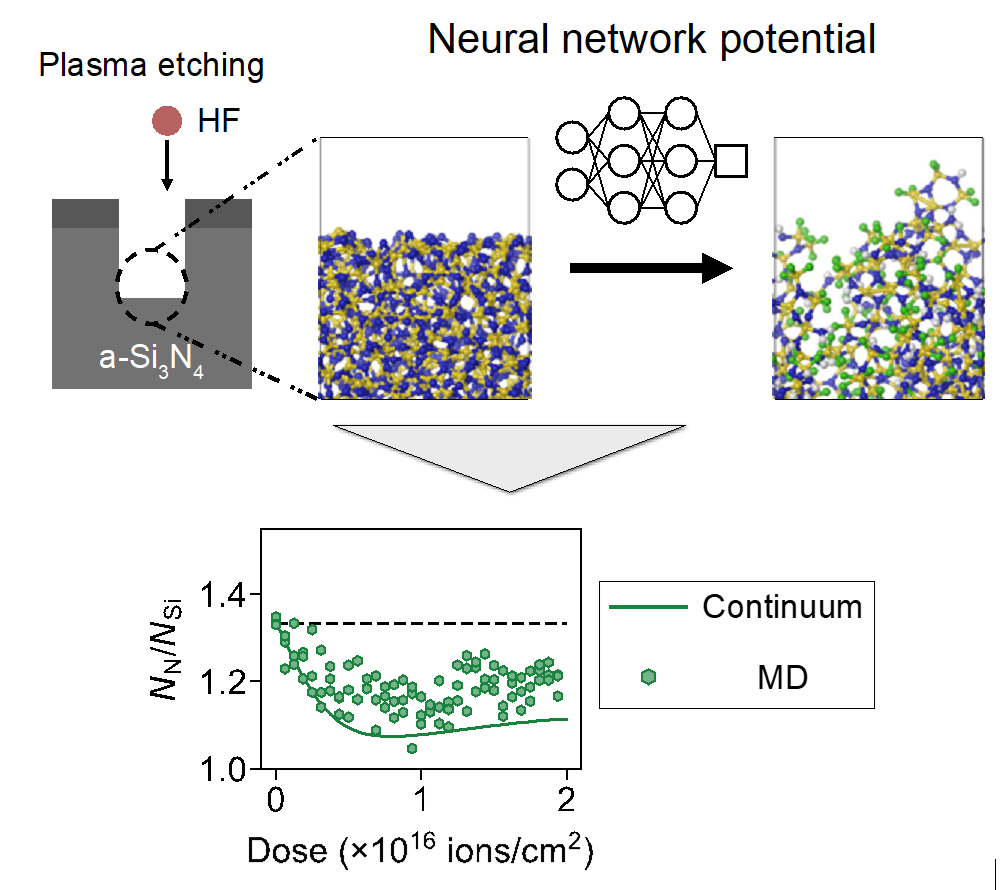
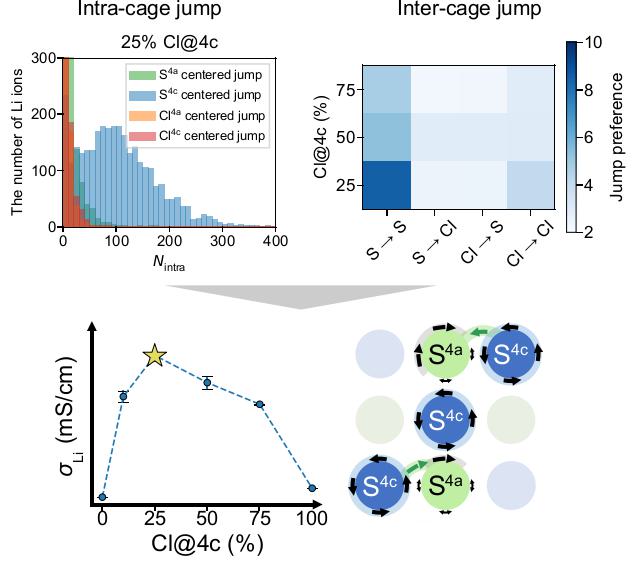
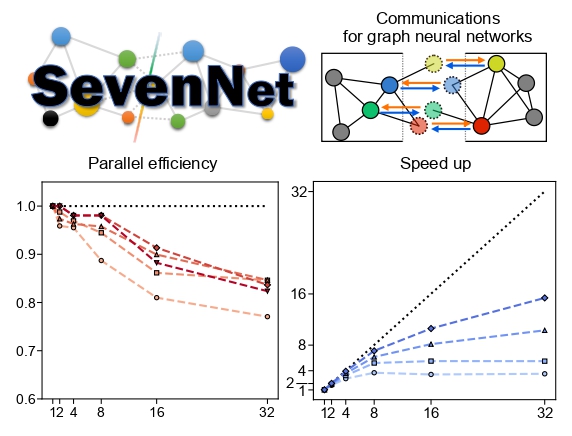
Machine learning potentials (MLPs) offer an efficient way to replicate potential energy surfaces
(PES) derived from first-principles calculations, significantly lowering computational costs.
However, generating high-quality training datasets remains expensive, particularly for
applications requiring high-level theoretical accuracy. Here, we introduce a cost-aware
multifidelity training strategy for MLPs that allows simultaneous utilization of low- and highfidelity datasets. Using an equivariant graph neural network as the underlying MLP architecture,
we validate this method across organic molecules, inorganic solids, and pretrained universal
MLPs. Our findings reveal that abundant, correlated data from low-fidelity sources can
effectively complement sparse high-fidelity datasets, bridging gaps in configurational and
compositional coverage. By strategically subsampling low-fidelity configurations, highfidelity PES can be reproduced accurately with minimal computational effort. Furthermore,
this approach addresses limitations associated with conventional methods for high-fidelity
MLPs, such as Δ-learning and transfer learning. This study was published in Journal of the
American Chemical Society 2025, 147, 1042.